In this session, we will perform reference-independent modification calling
on a dataset of human DNA that naturally contains cytosines modified due to methylation.
We will use the basecaller dorado that can perform basecalling and modification
calling when we issue a single command.
We will follow the pipeline illustrated below and we will execute all the steps
in this session.
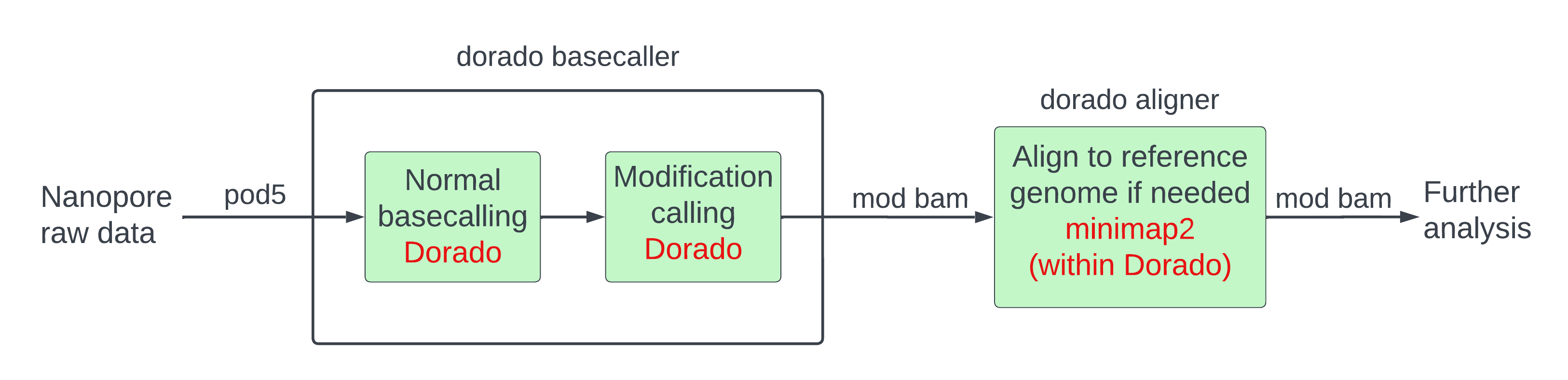
As you can see from the figure, there are a few differences compared to the pipeline we followed for the yeast dataset:
doradoperforms the job ofDNAscent(modification calling) in thedorado basecallercommand and callsminimap2(alignment) in thedorado alignercommand. Dorado can call methylation modification without relying on external software, whereas for custom modifications like BrdU, we have to use external software like DNAscent.- Modification calling and basecalling are performed with a single
dorado basecallercommand. - Modification calling is still bolted onto the output of basecalling.
- The workflow is reference-unanchored, so alignment is optional and follows modification calling.
Alignment is done through the
dorado alignercommand although it is just a different name forminimap2which is doing the job under the hood. The input to the alignment step is a mod BAM file and the output is another mod BAM file.
As the final result of our pipeline is a mod BAM file, all the tools that we have discussed
so far that work on mod BAM files carry over.
If you stop the analysis before the optional alignment step, then the unaligned mod BAM file
can still be used as input to modkit, samtools etc. but you cannot use these files in any
reference-dependent analysis such as IGV visualization, subsetting by region in samtools etc.
In this session, we will run the pipeline, get the final mod BAM file, and then you can explore the file yourself using the tools we have discussed thus far in the course or do a guided exploration by trying to solve one or more of the exercises at the end of the session.
We will be using a subset of the ‘Cliveome’ dataset released by Oxford NanoporeTech. The DNA is from the cells of the erstwhile CTO of ONT, Clive Brown, and we are re-creating a published pipeline.
We had performed a subset and some file conversions and calculations before the course began;
the steps are listed here and you can read them later.
We have already copied this data to the location ~/nanomod_course_data/human/
at the beginning of the course.
Inspect input data
We can inspect the pod5 file to see how many reads are on it.
input_pod5=~/nanomod_course_data/human/PAM63974_pass_58881fec_60.twenty_random_reads.pod5
pod5 inspect summary $input_pod5
There are 20 reads here. The output should look like the following.
File version in memory 0.3.2, read table version 3.
File version on disk 0.3.2.
File uses VBZ compression.
Batch 1, 20 reads
Found 1 batches, 20 reads
Basecalling and modification calling
Optional: Downloading dorado models
Downloading dorado models
We need to download the model dorado uses for basecalling and modification calling.
We will put them in a suitable directory.
dorado_model_dir=~/nanomod_course_references/dorado_models
model_config=dna_r10.4.1_e8.2_400bps_hac@v3.5.2
mkdir -p $dorado_model_dir
dorado download --model $model_config --directory $dorado_model_dir
model_config=dna_r10.4.1_e8.2_400bps_hac@v3.5.2_5mCG@v2
dorado download --model $model_config --directory $dorado_model_dir
We make a directory to store our modification calls.
output_dir=~/nanomod_course_outputs/human
mkdir -p $output_dir
We can basecall and modification call ten of our reads (-n 10) with
5mC methylation at CpG sites (with a suitable --modified-bases-models).
NOTE: the -b 10 -c 1000 are internal dorado parameters which we’ve chosen to fit
our virtual machines, and we are running in the slower CPU-only mode.
This step will take approximately ten minutes.
input_dir=~/nanomod_course_data/human
model_files=~/nanomod_course_references/dorado_models/dna_r10.4.1_e8.2_400bps_hac@v3.5.2
mod_model_files=~/nanomod_course_references/dorado_models/dna_r10.4.1_e8.2_400bps_hac@v3.5.2_5mCG@v2
output_mod_bam=~/nanomod_course_outputs/human/PAM63974_pass_58881fec_60.ten_reads.mod.bam
dorado basecaller $model_files $input_dir \
--verbose -n 10 -x cpu -b 10 -c 1000 --modified-bases-models $mod_model_files | \
samtools view --threads 8 -O BAM -o $output_mod_bam
Note down the program speed in number of samples per second.
Optional: modification calling speeds
Comparing our modification-calling speed with ONT’s benchmarks
You can compare the dorado basecalling rate we have recorded in samples per second to the ONT benchmarks listed here. Please note that some of ONT’s benchmarks were performed on expensive computers with many GPUs (see the cost tables at the link)! Our virtual machines do not have GPUs to minimize costs.
Optional: generating sequencing summary files
Dorado pipelines produce only BAM files but no fastq or sequencing summary files
You will see that this pipeline produces the mod BAM file directly
and no other file. If we perform alignment after modification calling,
as we shall do in a few moments, we will get another mod BAM file.
We do not get a fastq file or a sequencing summary file as we had
with the yeast pipeline.
We can generate a sequencing summary file if we wish to using the
dorado summary command as shown below.
This file is useful if we want to run tools like pycoQC.
input_mod_bam=~/nanomod_course_outputs/human/PAM63974_pass_58881fec_60.ten_reads.mod.bam
output_summ_file=~/nanomod_course_outputs/human/PAM63974_pass_58881fec_60.ten_reads.summary.txt
dorado summary $input_mod_bam > $output_summ_file
You can get a fastq file if you want using the command
samtools bam2fq $input_mod_bam > $output_fastq but we will not be doing it here.
Optional: multiple modification detection
Dorado can detect multiple types of methylation
We are not going to call both 5hmC and 5mC methylation here, but you can use the flag
--modified-bases 5mCG_5hmCG to do so.
Inspect results of modification calling
We can view the output modification data using nanalogue.
input_mod_bam=~/nanomod_course_outputs/human/PAM63974_pass_58881fec_60.ten_reads.mod.bam
nanalogue read-info $input_mod_bam
You can view a few reads and their mod counts above. You should see that all the reads are unmapped, but there are mod counts associated with each read. That is because we have just performed reference-independent modification calling.
Perform alignment post-modification calling
Optional: downloading the reference genome
Download the reference genome
We need to download a reference human genome which is a few GB. Please use the commands below.
cd ~/nanomod_course_references
url=https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids
wget "$url"/GCA_000001405.15_GRCh38_full_analysis_set.fna.fai
wget "$url"/GCA_000001405.15_GRCh38_full_analysis_set.fna.gz
gunzip GCA_000001405.15_GRCh38_full_analysis_set.fna.gz
Perform alignment
We perform alignment using the dorado aligner command which just runs minimap2 under the hood.
The -t 8 flag asks dorado to use 8 computational threads.
reference=~/nanomod_course_references/GCA_000001405.15_GRCh38_full_analysis_set.fna
cd ~/nanomod_course_outputs/human/
input_bam=PAM63974_pass_58881fec_60.ten_reads.mod.bam
output_bam=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.bam
dorado aligner -t 8 $reference $input_bam > $output_bam
Let us sort and index the output bam file
cd ~/nanomod_course_outputs/human/
input_bam=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.bam
output_bam=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.sorted.bam
samtools sort -o $output_bam $input_bam
samtools index $output_bam
Inspect alignment
We can use modkit to convert the output aligned file to TSV and then inspect it.
cd ~/nanomod_course_outputs/human/
input_mod_bam=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.sorted.bam
output_tsv=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.sorted.bam.tsv
modkit extract full $input_mod_bam $output_tsv
You can view a few rows of the $output_tsv file above.
As we’ve discussed in the session
on modification detection in yeast, the most important columns are read_id,
forward_read_position, ref_position, and mod_qual.
You can look up the full list of columns in the official documentation
here.
Do you see anything interesting in the query_kmer column?
The middle of the k-mer is the base of interest on
which modification has been called.
What base follows the cytosine of interest?
We can also inspect the file using the samtools and bedtools commands
we have encountered throughout the course.
cd ~/nanomod_course_outputs/human/
input_mod_bam=PAM63974_pass_58881fec_60.ten_reads.aligned.mod.sorted.bam
samtools view -c $input_mod_bam # count number of reads
samtools view -c --exclude-flags SECONDARY,SUPPLEMENTARY\
$input_mod_bam # count number of primary reads
bedtools bamtobed -i $input_mod_bam # inspect alignment coordinates
Optional: Quality control
Perform quality control with pycoQC
Quality control on the yeast dataset was covered in an optional part of a previous session; you can do that part first if you wish.
Similar to that lesson, if we run quality control on the ten-read alignment file we have
been making, we will get warnings and poor statistics.
So, we have run pycoQC ourselves on the entire Cliveome dataset
and have given you the resultant reports.
Please go to the directory ~/nanomod_course_data/human and open
the analysis.html file and have a look.
For your reference, we mention the pycoQC command used to generate these reports below
# DO NOT RUN THESE COMMANDS!
# They are for your reference only.
# To run them, you need to download and prepare the 'Cliveome'
# dataset as described in the dataset preparation page
# (to access this page, go to Home and look for the link
# Datasets under 'For Students').
# You can do so on your own computer (not the virtual machines
# used in the course) if you want to.
# For now, we just give you the results of the pycoQC step.
cd ~/nanomod_course_data/human/
input_dir= # this is an input directory on a different computer
seq_summ="$input_dir"/cliveome_kit14_2022.05/gdna/flowcells/ONLA29134/20220510_1127_5H_PAM63974_a5e7a202/sequencing_summary_PAM63974_58881fec.txt
alignment_bam="$input_dir"/cliveome_kit14_2022.05/gdna/basecalls/PAM63974/bonito_calls.bam
pycoQC -f $seq_summ -a $alignment_bam -o ./analysis.html -j ./analysis.json
Further analysis
For further analysis, we have prepared a subset of the Cliveome dataset by choosing
reads passing through a specific region of the human genome.
It is located at ~/nanomod_course_data/human/bonito_calls.subset.sorted.bam.
Although this file’s name ends in .bam, it is a mod BAM file.
So all the downstream analysis and visualizations tools we have discussed thus far in
the course can be used to analyse and visualize this file respectively.
You can now explore the BAM file yourself using the tools we have talked about so far. Or you can do one or some of the exercises below to explore the BAM file in a guided way.